
The laboratory, directed by Dr. Scott Kern, is constructing a molecular genetic foundation for understanding pancreatic cancer. In the past, pancreatic cancer has advanced from being one of the most difficult research subjects, for which little genetic understanding was available, to become one of the best-described genetic models of a human cancer type. The laboratory and its collaborators aim to use this new understanding to design gene-based tests for early detection and to develop strategies for effective therapy.
So, what does an individual pancreatic cancer look like when we view it with the lenses provided by the molecular genetic technology? Basically, it's a genetic mess. But it's a mess with underlying patterns, which they are now beginning to unravel. You can visualize the patterns through a brief tour of the Allelotype or of the Cell Line Mutation Profiles below or review the roles of individual genes and chromosomes in pancreatic cancer.
Pancreatic cancer was shown to be distinctively different from another well-studied gastrointestinal cancer type, colon cancer. APC gene mutations, seen in most colon neoplasms, are not found in pancreatic cancer. Also, a DNA mismatch repair defect, seen in 15% of colorectal cancer, is uncommon in pancreatic cancer. This research group therefore does not see pancreatic cancer as being modeled after any other cancer type. It is its own, very characteristic, entity. The aggressiveness of the disease exceeds that of most other carcinomas. Chemotherapeutic agents which may be active against other malignancies do not work effectively when used for pancreatic cancer. The laboratory therefore pursues a focused attempt to study pancreatic cancer as comprehensively and efficiently as can be achieved.
The Discoveries
The Kern laboratory has defined the frequency and positions of deletions of chromosomal material, and discovered highly frequent mutational changes affecting the DPC4 gene, p16 gene, and p53 gene. K-ras gene mutations are known to be common in pancreatic cancer. Mutant K-ras genes were present in the stool samples of patients with either pancreatic cancer or the precursor lesions for the cancer. A genetic link with familial breast cancer was discovered. Their work has suggested a model for the biology of pancreatic cancer, shown in the Mutations and the Cell Cycle below.
Mutations and the Cell Cycle ▼
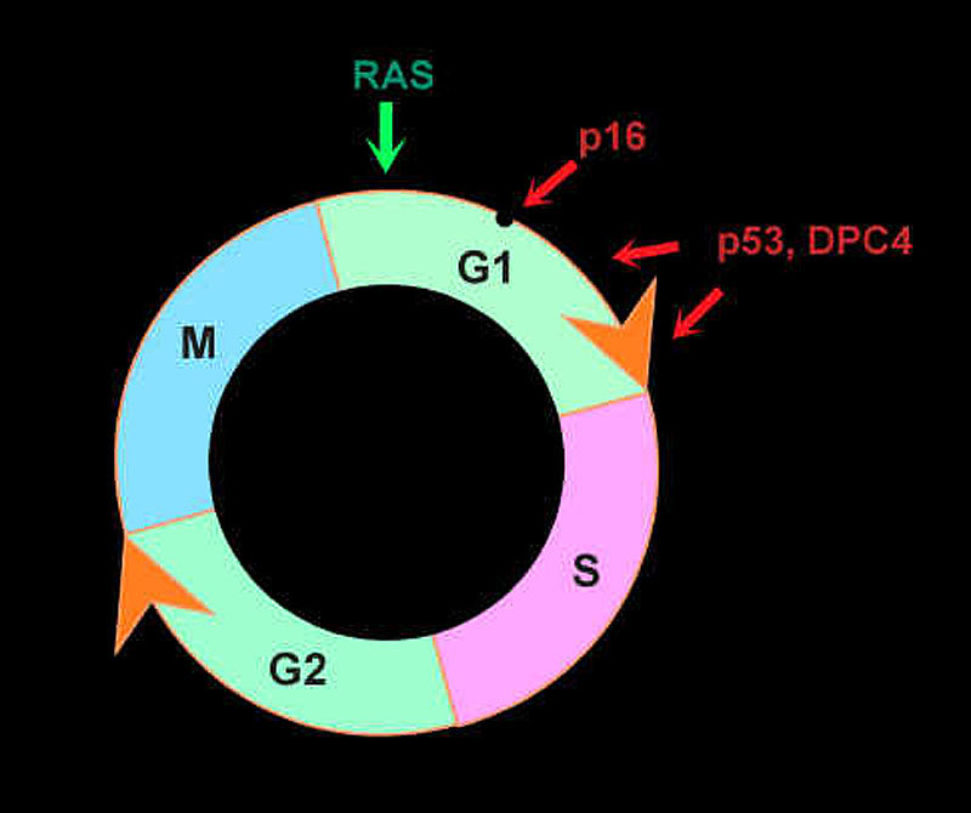
In this figure, the genetic changes of pancreatic cancer are shown in a theoretical framework, emphasizing the disruption of cell cycle controls. Ras mutations provide an inappropriate stimulus for cells to grow in a phase called G1. The p16 and p53 genes act at multiple checkpoint positions to restrict inappropriate G1 progression. DPC4 mutations probably affect this phase as well.
The Allelotype of Pancreatic Cancer ▼
In this figure, the genetic changes of pancreatic cancer are shown in a theoretical framework, emphasizing the disruption of cell cycle controls. Ras mutations provide an inappropriate stimulus for cells to grow in a phase called G1. The p16 and p53 genes act at multiple checkpoint positions to restrict inappropriate G1 progression. DPC4 mutations probably affect this phase as well.
Cell Line Mutation Profiles ▼
K-Ras Mutations ▼
Why are K-ras mutations important?
Mutations of the K-Ras gene occur in over 90% of pancreatic carcinomas. No other human tumor comes close in mutational frequency. The ras pathway is important in the transmission of growth-promoting signals from the cell surface receptors, eventually toward the nucleus where these signals affect the production and regulation of other key proteins. It is interesting that although most mutations in genes are expected to cause their inactivation, with the Ras genes the opposite happens - they become more active in signaling. This is because of the engineering design of the protein. The ras signal is turned off by a molecular switch, which depends on an enzyme activity. In picturesque terms, the nucleotide GTP (guanidine triphosphate) engages the switch to keep it in the "on" state. A portion of the Ras protein has an enzyme activity (a GTPase) which cleaves the GTP. This turns the switch "off" after the brief "on" period. In reality, the mutations of Ras do indeed inactivate a function, as most mutations are expected to do. The GTPase is inactivated by the mutations. But this now means that GTP continues to engage the switch, and the Ras signaling function is unable to be turned "off". A number of drug companies are now investigating ways to abate the signaling function of Ras, which could provide a rational therapy for pancreatic cancer.
Where do they occur?
Ras mutations involve only certain amino acids, those which interfere with the GTPase function. In pancreatic cancer, mutations are essentially seen only at the twelfth position, (codon or amino acid 12), with rare exceptions seen at codon 13. Most mutations in pancreatic cancer change a glycine at codon 12 to a valine or aspartate. The mutation to serine is quite unusual in pancreatic cancer, a peculiar finding since it is a common mutation in other tumor types which have K-ras mutations.
When do they occur?
It is now known that K-ras mutations occur long before the formation of the actual cancer. They form in the precancerous stages, within lesions termed PIN, or Pancreatic Intraepithelial Neoplasia. These are ducts with clones of early neoplastic cells which have not yet invaded through the duct wall. In reality, these lesions are among the most common neoplasms of humans, occurring in nearly a third of elderly people. They are often unfortunately called "hyperplasia", a term which implies the lack of neoplastic character. Indeed, a rough estimate suggests that less than 1% of the lesions ever become invasive, which is to say, become a cancer. Thus, in many respects, they are similar to the adenomas of the colon; nearly a third of people will develop adenomas, yet only a small proportion will progress to colon cancer. A major effort of the Kern laboratory is to determine which of the genetic changes in PIN mark the lesions at high risk for invasion. K-ras mutations are uncommon in the early forms of PIN, but are found in the majority of advanced lesions. PIN having K-ras mutations will probably develop in 10% of persons during life. Thus, K-ras mutations are still not the specific marker of high-risk lesions that they would like to identify for diagnostic purposes. It should be obvious that a major goal is the diagnosis of pancreatic neoplasms at the earliest possible date, preferably in the advanced PIN lesions and prior to the formation of frank cancer which invades through the duct wall and spreads to other organs of the patient.
These studies are done in collaboration with Dr. Ralph Hruban and his efforts to study K-Ras mutations.
p53 Mutations ▼
Why are p53 mutations important?
The p53 gene suffers deletions in nearly 90% of pancreatic cancers. Nearly 60-70% have a point mutation which inactivates the remaining copy of the gene. Among the various human tumor types, p53 mutations are the most common known. p53 is thought to be a master controller of certain tumor-suppressive pathways. It binds specific sequences of DNA and directs the production of RNA from nearby genes, a process called transcriptional activation. Because all naturally-occurring p53 mutations impair p53's ability to bind DNA, this important tumor-suppressive function is lost.
Any clues to why they occur?
In comparison with other tumor types, there is a peculiar preference for pancreatic carcinomas to have intragenic deletions, and in particular 1-2 base pair microdeletions (17% versus, for example, less than 1% in colon cancer). These deletions frequently were within repeated runs of single nucleotides or nucleotide pairs. The cause is unclear, but the possibilities are intriguing, since such mutations are of the type seen with exposure to mutation-inducing chemicals. As a bit of background, it has been suggested that exposure to particular environmental or endogenous mutagens might be related to specific mutation spectra in p53. For example, aflatoxin-associated mutations in hepatocellular carcinoma very frequently involve a particular site within the p53 gene. It has also been observed that different mutations are favored in colorectal, lung or UV-induced skin carcinomas. Knowledge of the causative agents or the mechanisms of genetic alteration might provide clues to the processes involved in causing pancreatic cancers. One would hope to use this understanding to help reduce the incidence of the disease among the population.
p16 Mutations ▼
Why are p16 mutations important?
Mutations of the p16 gene occur in pancreatic cancer at a rate higher than that reported in any other tumor type. Nearly 90% of tumors lose one copy (termed loss of heterozygosity, or LOH) by deletion. In 40% of cases, the other copy is also lost (termed a homozygous deletion, or HD). In another 40%, a small mutation in the p16 gene inactivates its function. The remainder of tumors usually turn off the p16 gene by a process called methylation that, strictly speaking, is not a form of mutation. p16 is one of the inhibitors for enzymes (termed cyclin-dependent kinases, or CDKs) which drive a cell's progression through the division cycle.
Wasn't there a controversy?
Initial reports of mutational analyses of p16 in human tumours were controversial, and it was suggested that mutations in the p16 gene might be selected by or induced upon tissue culture. This would make it an artifact unrelated to human tumorigenesis. The first convincing evidence for human tumor acquiring mutations at a high rate came through the mutational screen for pancreatic carcinoma. Mutations and deletions, performed on pancreatic cancer xenografts, were confirmed in all available original primary tumours. Also, comparisons of parallel xenografts, where multiple grafts were taken from the same primary tumours, showed consistent results, thus excluding any in vitro artifacts. p16 gene mutations were variably reported in esophageal squamous cell carcinoma and in the germline of some patients with familial melanoma, but were largely absent in a wide variety of other tumours that exhibit a high frequency of 9p loss. In part, it turns out that p16 sustains deletions of both copies, and these changes were initially missed since this type of mutation is difficult to see when geneticists examine human tumors directly. p16 inactivations are now thought to be nearly as common as p53 mutations in multiple types of human cancer.
Can I get familial pancreatic cancer from a p16 mutation?
There is a syndrome called familial atypical mole/melanoma, which is due to p16 mutations that are passed on within a family from parent to child. The risk of pancreatic cancer is higher within these families. But to date, all pancreas cancer families reported to have an inherited p16 mutation have also had melanomas within the family. Consult your doctor if you think your family might have this syndrome, since testing is available to identify those family members carrying the mutant p16 gene. Knowing who is at risk can allow them to take preventative action and to attempt to identify skin lesions at an early, curable stage.
What about p15?
There is at least one other candidate tumour-suppressor gene, p15, located near p16. Because the homozygous deletions involving p16 frequently extend into distant flanking markers, p15 is known to be involved as well by those homozygous deletions of 9p in pancreatic carcinoma. But p15 is not mutated in pancreatic cancer, and therefore there is no direct evidence of p15 being a tumor-suppressor gene in this tumor type.
DPC4 Mutations ▼

Why did you go looking for this gene?
The nearly ubiquitous deletions (termed loss of heterozygosity, or LOH) of chromosome 18q suggested the inactivation of a very important tumor-suppressor gene which resides there, the identity of which was unknown at the time. 18q is one of the most frequently deleted chromosomal arms among a variety of human cancer types. A candidate gene on 18q, the DCC gene, had not been reported to be mutated in pancreatic cancer. Thus, for years, the search continued for the elusive gene.
How did you find it?
The laboratory decided to scan the suspected area with multiple markers, in order to evaluate the integrity of the chromosome. In one small area (about 1% of the length of the chromosome, in the region termed 18q21.1), they found multiple small deletions of a type which were previously thought to be quite rare. These were deletions which affected both normal copies of the chromosome, termed "homozygous deletions", and they were found in 30% of the cancers. A gene was cloned from this region. Because this was the fourth locus to be investigated for this special form of deletion in pancreatic cancer, they named the gene DPC4 (for Deleted in Pancreatic Cancer, locus 4). In a number of cases which did not have these homozygous deletions, other forms of mutations were found which would inactivate this gene. In all, 50% of pancreatic cancers, as well as lesser proportions of other cancers (such as bladder, breast, lung, bile duct, and colon tumors), experience a total inactivation of the DPC4 gene due to mutations. The gene is suspected of normally acting to prevent the occurrence of tumors of the pancreas and other organs, and so is one of a small number of known tumor-suppressor genes.
What does DPC4 do?
A fascinating clue to the biochemical function of this protein was suggested by an Internet-based computer search of DNA databases. DPC4 is very similar to a gene in the fruit fly, Drosophila malanogaster, which is needed in early fly development. The signals are part of the TGF-ß superfamily of signaling pathways. When we used a genetic strategy to knock out the TGF-ß gene in a cancer cell, we found we had also inactivated the TGF-ß pathway. These pathways usually act to promote differentiation (maturation to normal adult forms) and to slow the growth of cells. Similar proteins are also found in the worm C. elegans. We have recently found that DPC4 binds to a specific sequence in DNA, allowing DPC4 to turn on the expression of certain genes.
What does TGF-ß do?
TGF-ß has a role in humans similar to that in other species, and most normal cells stop proliferating when exposed to TGF-ß. One of the most remarkable findings about tumor cells from any of a number of sites in the body is that the majority of them do not become suppressed by TGF-ß. It is therefore hoped that the finding of mutations in DPC4 will help us to understand one of the most basic abnormalities of cancers, which is their inability to respond to the body's own normal signals which should control their growth. Perhaps by finding a way to overcome this blockade to TGF-ß signals, we can re-establish a natural and powerful means to rein in the tragic aggressiveness of cancer.
MKK4 Mutations ▼
Why are MKK4 mutations important?
Inactivation of the MKK4 gene occurs in pancreatic cancer at a low rate, in about 4% of cases. But this is a new and unexpected type of tumor-suppressor pathway, and it may have a lot to teach us. MKK4 is part of a signaling system that is stimulated in the presence of stress to the cell (including chemotherapy), and it may control differentiation and programmed cell death - termed apoptosis. Once we know the roles of the other members of the pathway, we may find more mutations in other genes.
TGF-ß Receptor and Activin Receptor Mutations ▼
What does TGF-ß do?
TGF-ß is a protein that is made and released by cells, so it is present in all tissues and even in blood. Most normal cells stop proliferating when exposed to higher levels of TGF-ß. One of the most remarkable findings about tumor cells from any of a number of sites in the body is that the majority of them do not become suppressed by TGF-ß. When we study the problems of the TGF-ß system in cancers, we are trying to understand one of the most basic abnormalities of cancers, which is their inability to respond to the body's own normal signals which should control their growth. Perhaps by finding a way to restore these TGF-ß signals, we can re-establish a natural and powerful means to rein in the tragic aggressiveness of cancer.
Why are TGF-ß receptors important?
Cells have receptors on their surface that are specially made to send signals through the cell when the cell comes in contact with certain kinds of molecules. TGF-ß receptors have this role. They send signals through certain pathways in the cell. One of the most important pathways includes the protein Dpc4, which is often mutated in pancreatic cancer. Mutations or deletions of the TGF-ß receptors is thus another way that cancer cells become resistant to the normal growth controls that operate in the body. There are two types of TGF-ß receptor, called the type I receptor and the type II receptor. About 2% of pancreatic cancer have mutations of each receptor type. We were the first to find mutations of the type II receptor in pancreatic cancer, and the first to find genetic inactivation of the type II receptor in any cancer type.
What about activin?
There are other proteins similar to TGF-ß, and these other proteins need their own receptor system as well. One of these is called activin. Not much is known about activin in cancers, but it appears to suppress cell growth just as TGF-ß does. Most of the attention, however, had focused on TGF-ß. We decided to take a look at the activin system, and were the first to find mutations of the activin type 1B receptor in human cancer. About 2% of pancreas cancers have genetic inactivation of the activin type 1B receptor, which would be expected to prevent activin from serving its role to control the number of cancer cells.
BRCA2 ▼
This is a breast cancer gene, right?
Yes and no. The laboratory used RDA to identify a homozygous deletion in a pancreatic cancer, and this deletion happened to include the location of the BRCA2 gene that was being sought as a cause of familial breast cancer. Aided by the localization available from this pancreatic cancer, the BRCA2 was then found by other investigators. The laboratory then searched for mutations of BRCA2 and found them in nearly 7% of sporadic pancreatic cancers. It seems that breast, ovarian, and pancreatic cancers can all occur at higher rates in families that inherit a mutation in the BRCA2 gene. Some of these families nonetheless have very few cases of cancer, and without the benefit of genetic studies, the pattern of cancer will not appear obviously familial, but will seem to be sporadic.
LKB1/STK11 - The Peutz-Jeghers Gene ▼
This refers to a syndrome that runs in families, right?
Yes, early in this century a physician named Dr. Peutz described patients who had multiple polyps of their intestinal tract. Later, Dr. Jeghers reported additional cases, some of them studied here at Johns Hopkins. In the late 1980s, Dr. Giardiello of Hopkins reported that persons with this syndrome were at a high risk of developing pancreatic cancer. In 1999, the Kern Laboratory, Dr. Hruban, and collaborators in The Netherlands worked together to find the proof of this association. The patients with this syndrome were known to have one mutant copy of the LKB1/STK11 gene. The Hopkins group found that in one of these patients' cancers, the remaining normal copy of the gene was lost. They then also showed that about 5% of pancreatic and biliary cancers from patients without this syndrome, also had mutations or deletions of this gene. This gene is therefore acting to prevent the emergence of pancreatic cancer, and genetic alterations of the gene occur in thousands of pancreatic cancers each year.
Telomeric and Mitochondrial DNA ▼
Telomeres do what?
Telomeres are the ends of chromosomes. They are like caps on a fence post, special structures that keep chromosomes from falling apart; when that happens, the chromosome can lose (delete) genes from region, or the broken end can join with other chromosomes to form a translocation. We have found that virtually all of the early forms of pancreatic neoplasia, the precursors to cancer, have dramatically short telomeres. We believe that this is the underlying reason for many of the genetic changes in pancreatic ducts, changes that over a period of decades can lead to the evolution of a cancer. Without the loss of so much telomeric DNA, it is possible that people would never get pancreatic cancer.
What do mitochondria have to do with cancer?
The answer to this question may be a surprise. Mitochondria are the energy powerhouses of the cell, in which oxygen is consumed to produce chemical power for other cell functions. The mitochondria are not in the cell nucleus where most cellular DNA resides, and mitochondria indeed have their own separate DNA. The vast majority of pancreatic cancers have mutations in mitochondrial DNA. But these mutations seldom seem to have an effect on normal mitochondrial functions. Such mutations may just be a fairly normal accompaniment of aging. We don't yet know whether mitochondria have any special role in helping to produce cancers. But because pancreatic cancer cells have such a dramatic increase in the amount of mitochondrial DNA compared to normal cells, it may in the future be easier to detect cancer by detection of the mitochondrial mutations than by detection of mutations in the nuclear DNA.
Various Chromosomes ▼
1p, 3p, 6p & q, 8p, 10q, 12q, 13q, 18p, 21q, 22q
A roadmap to future discoveries.
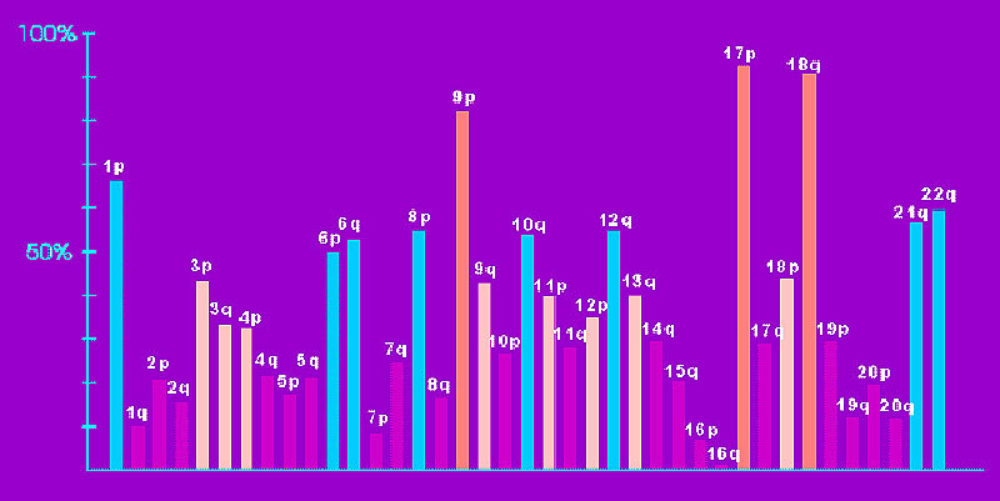
The allelotype showed us a large number of chromosomal sites that are deleted in pancreatic cancer. Some of these are known to have mutant genes. For example, the deletions of 17p (the short arm of chromosome 17) help to inactivate the p53 and MKK4 genes that reside there. The p16 gene is on 9p, the DPC4 gene on 18q. But there are other chromosomal arms which are not deleted quite so universally, but for which losses have been seen in 40-60% of pancreatic cancers. These sites are listed above, and are also suspected of harboring tumor-suppressor genes. Because of the large number of these sites, it is likely that they play an important role in determining the tumor's aggressive behavior. Some are described in more detail:
1p - A region of the short arm of chromosome 1 is lost in nearly 50% of the cancers. The p18 gene, which encodes a cyclin-dependent kinase inhibitor highly similar to p16, is located in this region of 1p, but is not mutated in the cancers; thus, the tumor-suppressor gene on 1p remains unknown.
8p - A region of the short arm of chromosome 8 is lost in nearly 50% of the cancers. In one region of this chromosome, a cell line is known that has deletions of both copies, but it is not yet clear that there is a tumor-suppressor gene located there.
18p - The losses of 18p probably occur because of the loss of an entire copy of chromosome 18, including both the short (p) and long (q) arms. Since the laboratory has identified a major tumor-suppressor gene (DPC4) on the q arm, they view the loss of the p arm as a "passenger mutation", innocently carried along by the losses which inactivate one or more genes on 18q. Both copies of the DCC gene is deleted in a few pancreatic cancers, but DCC is a such a large gene that it is difficult to study.
22q - A tumor-suppressor gene termed Schwannomin (SCH) is located on chromosome 22q, and is mutated in neurofibromatosis type 2. They found no mutations of SCH in pancreatic cancer. Again, another tumor-suppressor gene probably lies on the chromosome and is waiting to be discovered.
FISH Picture of Pancreatic Cancer ▼
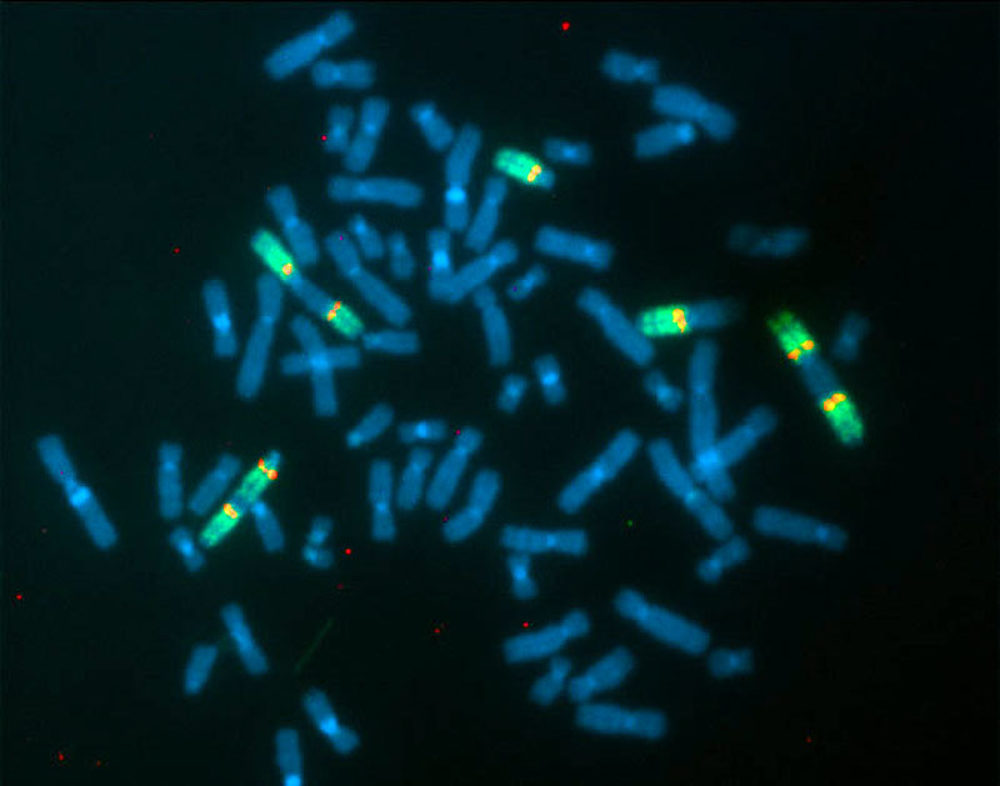
Fluorescent In-Situ Hybridization (FISH) using a chromosomal paint for chromosome 13 (in green) and a probe from near the BRCA2 gene (in red), applied to a metaphase spread from the pancreatic cancer cell line Colo357. This cell line has a translocation near, but not exactly at, the BRCA2 gene. If you look carefully, you will see green color extends on both sides of the red dots, indicating that the BRCA2 gene is entirely contained within an undisturbed portion of the chromosome 13. The multiple forms of chromosome 13, including both 'head to head' and 'head to tail' rearrangements which produce a double version of the chromosome, as well as some normal copies of the chromosome, illustrate well the bizarre chromosomal rearrangements which can be seen in pancreatic cancer. Note also the eight copies of the chromosome instead of the normal two. This is one of the manifestations of the property of cancer cells called 'aneuploidy', where the number and structure of the chromosomes is altered.
Bibliography
THESE BIBLIO SETS ARE VERY OLD
Original Research Reports ▼
- Seymour AB, Hruban RH, Redston MS, Caldas C, Powell SM, Kinzler KW, Yeo CH, Kern SE. Allelotype of pancreatic adenocarcinoma. Cancer Res 1994; 54:2761-2764.
- Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CH, Kern, SE. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994; 54:3025-3033.
- Caldas C, Hahn SA, Hruban RH, Redston MS, Yeo CJ, Kern, SE. Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res 1994; 54:3568-3573.
- Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nature Genetics 1994; 8:27-32.
- Schutte M, da Costa LT, Hahn SA, Moskaluk C, Hoque ATMS, Rozenblum E, Weinstein CL, Bittner M, Meltzer PS, Trent JM, Yeo CJ, Hruban RH, Kern SE. A homozygous deletion identified by representational difference analysis in pancreatic carcinoma overlaps the BRCA2 region. Proc Natl Acad Sci USA 1995; 92:5950-5954.
- DiGiuseppe, Redston MS, Yeo CJ, Kern SE, Hruban RH. p53-independent expression of the cyclin-dependent kinase inhibitor p21 in pancreatic carcinoma. Amer J Pathol 1995; 147:884-888.
- Day JD, DiGiuseppe JA, Yeo CJ, Lai-Goldman M, Anderson SM, Kern SE, Hruban RH. Immunohistochemical evaluation of Her-2/neu oncogene expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasms. Human Pathology 1996; 27:119-124.
- Hahn SA, Seymour AB, Hoque ATMS, Schutte M, da Costa LT, Redston MS, Caldas C, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. Allelotype of pancreatic adenocarcinoma using a xenograft model. Cancer Res 1995; 55: 4670-4675.
- Schutte M, Rozenblum E, Moskaluk CA, Guan X, Hoque ATMS, Hahn SA, da Costa LT, de Jong PJ, Kern, SE. An integrated high-resolution physical map of the DPC/BRCA2 region at chromosome 13q12-13. Cancer Res 1995; 55:4570-4574.
- Rozenblum E, Schutte M, Kern SE. INK4 genes in pancreatic carcinoma. Oncology Reports 1996; 3:743-745.
- Hahn SA, Schutte M, Hoque ATMS, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor-suppressor gene at human chromosome 18q21.1. Science 1996; 271:350-353. [8th most cited research paper of 1996, Science Watch 1997; 8:2]
- Hoque ATMS, Hahn SA, Schutte M, Kern SE. DPC4 gene mutation in colitis-associated neoplasia. Gut 1997; 40:120-122.
- Hahn SA, Hoque ATMS, Moskaluk CA, da Costa LT, Schutte M, Rozenblum E, Seymour AB, Weinstein CL, Yeo CJ, Hruban RH, Kern SE. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res 1996; 56:490-494.
- Brat DJ, Hahn SA, Griffin CA, Yeo CJ, Kern SE, Hruban RH. The structural basis of molecular genetic deletions: An integration of classical cytogenetic and molecular analyses in pancreatic adenocarcinoma. Am J Pathol 1997; 150:383-391.
- Moskaluk CA, Hruban RH, Lietman A, Smyrk T, Fusaro L, Fusaro R, Lynch J, Yeo CJ, Jackson CE, Lynch HT, Kern SE. Novel p16INK4A mutation in familial pancreatic carcinoma. Human Mut 1998 (in press).
- Schutte M, Hruban RH, Hedrick L, Molnar’Nadasdy G, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero R, Meltzer PS, Hahn SA, Kern SE. DPC4 in various tumor types. Cancer Res 1996, 56:2527-2530.
- Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JKV, Markowitz S, Hamilton SR, Kern SE, Kinzler K, Vogelstein B. Evaluation of candidate tumor-suppressor genes on chromosome 18q in colorectal cancers. Nature Genet 1996; 13:343-346.
- Riggins GJ, Thiagalingam S, Rozenblum E, Weinstein CL, Kern SE, Hamilton SR, Willson JKV, Markowitz SD, Kinzler KW, Vogelstein B. MAD-related genes in the human. Nature Genetics 1996; 13:347-349.
- Derynck R, Gelbart WM, Harland RM, Heldin C-H, Kern SE, Massagué J, Melton DA, Mlodzik M, Padgett RW, Roberts AB, Smith J, Thomsen GH, Vogelstein B, Wang X-F. Nomenclature: vertebrate mediators of TGFb family signals. Cell 1996; 87:173.
- Moskaluk C, Hruban RH, Schutte M, Lietman AS, Smyrk T, Fusaro L, Rusar R, Lynch J, Yeo CJ, Jackson CE, Lynch HT, Kern SE. Genomic sequencing of DPC4 in the analysis of familial pancreatic cancer. Diag Mol Pathol 1997; 6:85-90.
- Barrett MT, Schutte M, Kern SE, Reid BJ. Allelic loss and mutational analysis of the DPC4 gene in esophageal adenocarcinoma. Cancer Res 1996; 56:4351-4353.
- Goggins M, Schutte M, Lu J, Moskaluk, CA, Weinstein C, Petersen G, Yeo CJ, Jackson, CE, Lynch HT, Hruban RH, Kern SE. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res 1996, 56:5360-5364.
- Rozenblum E, Schutte M, Goggins M, Hahn SA, Lu J, Panzer S, Zahurak M, Goodman SN, Hruban RH, Yeo CJ, Kern SE. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res 1997; 57:1731-1734.
- Moskaluk C, Kern SE. Microdissection and PCR amplification of genomic DNA from histologic tissue sections. Am J Pathol 1997; 150:1547-1552.
- Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res 1997; 57:2140-2143.
- Zhang L, Zhou W, Velculescu VE, Kern SE, Hruban RH, Hamilton SR, Vogelstein B, Kinzler KW. Gene expression profiles in normal and cancer cells. Science 1997; 276:1268-1272.
- Zhou W, Sokoll LJ, Bruzek DJ, Zhang L, Velculescu VE, Goldin SB, Hruban RH, Kern SE, Hamilton SR, Chan DW, Vogelstein B, Kinzler KW. Identifying markers for pancreatic cancer by gene expression analysis. Cancer Epid Bio Prev 1998; 7:109-112.
- Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff I, Schmiegel W, Baylin SB, Kern SE, Herman JG. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997; 57:3126-3130.
- Goggins M, Offerhaus GJA, Hilgers W, Griffin CA, Shekher M, Tang D, Sohn T, Yeo CJ, Kern SE, Hruban RH. Adenocarcinomas of the pancreas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology: poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol 1998; 152:1501-1507.
- Sirard C, de la Pompa JL, Elia AJ, Itie A, Mirtsos C, cheung A, Hahn S, Wakeham A, Schwartz L, Kern SE, Rossant J, Mak TW. The tumor suppressor gene Dpc4/Smad4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev 1998; 12:107-119.
- Okami K, Wu L, Riggins G, Cairns P, Goggins M, Evron E, Halachmi N, Ahrendt SA, Reed AL, Hilgers W, Kern SE, Sidransky D, Jen J. Analysis of PTEN/MMAC1 alterations in aerodigestive tract tumors. Cancer Res 1998; 58:509-511.
- Zhou S, Buckhaults P, Zawel L, Bunz F, Riggins G, Dai JL, Kern SE, Kinzler KW, Vogelstein B. Targeted deletion of Smad4 shows it is required for transforming growth factor-b and activin signaling in colorectal cancer cells. Proc Natl Acad Sci USA 1998 95:2412-2416.
- Zawel L, Dai J, Buckhaults P, Zhou S, Kinzler, KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Molec Cell 1998; 1:611-617.
- Goggins M, Lietman A, Miller RE, Yeo CJ, Jaffee E, Coleman J, O’Reilly S, Cullen B, Kern SE, Hruban RH. The pancreatic cancer web site at Johns Hopkins: patterns of use and benefits of an institutional disease-based web site. JAMA 1998; 280:1309-1310.
- Su GH, Hilgers W, Shekher M, Tang D, Yeo CJ, Hruban RH, Kern SE. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor-suppressor gene. Cancer Res 1998; 58:2339-2342.
- Wilentz RE, Geradts J, Offerhaus GHA, Kang M, Goggins M, Yeo, CJ, Kern SE, Hruban RH. Inactivation of the p16 (INK4A) tumor-suppressor gene early in pancreatic neoplasia: loss of intranuclear expression. Cancer Res 1998; 58:4740-4744.
- Dai JL, Turnacioglu K, Schutte M, Sugar AY, Kern SE. DPC4 transcriptional activation and dysfunction in cancer cells. Cancer Res 1998; 58:4592-4597.
- Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE. Genetic alterations of the TGFb receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res 1998; 58:5329-5332.
- Hilgers W, Tang DJ, Sugar AY, Shekher MC, Hruban RH, Kern SE. High-resolution deletion mapping of chromosome 1p in pancreatic cancer identifies a major consensus at 1p35. Genes Chromosom Can 1999; 24:351-355.
- DaDai JL, Bansal RK, Kern SE. G1 cell cycle arrest and apoptosis induction by nuclear Smad4/Dpc4 – phenotypes reversed by a tumorigenic mutation. Proc Natl Acad Sci USA 1999; 96:1427-1432i J, Schutte M, Sugar A, Kern SE. TGFb responsiveness in DPC4-null cancer cells. Molec Carcinog 1999; 26:37-43.
- Su GH, Hruban RH, Bova GS, Goggins M, Bansal RK, Tang DT, Shekher MC, Entius MM, Yeo CJ, Kern SE. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999; 154:1835-1840.
- Hilgers W, Su GH, Groot Koerkamp B, Tang DJ, Shekher MC, Sugar AY, Yeo CJ, Hruban RH, Kern SE. Novel homozygous deletions of chromosome 18q22 in pancreatic adenocarcinoma identified by STS marker scanning. Genes Chrom Can 1999; 25:370-375.
- Hilgers W, Koerkamp BG, Geradts J, Tang DJ, Yeo CJ, Hruban RH, Kern SE. Genomic FHIT alterations in RER+ and RER- adenocarcinomas of the pancreas. Genes Chrom Cancer 2000, 27:239-243.
- Hilgers W, Song JJ, Hayes M, Hruban RR, Kern SE, Fearon ER. Homozygous deletions inactivate DCC, but not MADH4/DPC4/SMAD4, in a subset of pancreatic and biliary cancers. Genes Chrom Cancer 2000; 27:353-357.
- Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, Yeo CJ, Kern SE, Hruban RH. Immunohistochemical labeling for Dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol 2000; 156:37-43.
- Wilentz RE, Goggins M, Redston M, Marcus VA, Sohn TA, Yeo CJ, Choti M, Zahurak M, Johnson K, Tascilar M, Offerhaus GJA, Hruban RH, Kern SE. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: A newly described and characterized entity. Am J Pathol 2000; 156:1641-1651.
- Goggins M, Hruban RH, Kern SE. The late temporal pattern of BRCA2 inactivation in pancreatic intraductal neoplasia: Evidence and implications. Am J Pathol 2000; 156:1767-1771.
- Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, Kern SE, Hruban RH. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res 2000; 60:2001-2005.
- Jones J, Kern SE. Functional mapping of the MH1 DNA-binding domain of DPC4/SMAD4. Nucl Acids Res 2000; 28:2363-2368.
- Iacobuzio-Donahue CA, Klimstra DS, Volkan Adsay N, Wilentz RE, Argani P, Sohn TA, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Dpc4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: comparison with conventional ductal carcinomas. Am J Pathol 2000; 157:755-761.
- Su GH, Sohn TA, Ryu B, Kern SE. A novel histone deacetylase inhibitor identified by high-throughput transcriptional screening of a compound library. Cancer Res 2000; 60:3137-3142
- Iacobuzio-Donahue CA, Wilentz RE, Argani P, Yeo CJ, Kern SE, Hruban RH. Dpc4 protein in mucinous cystic neoplasms of the pancreas: Frequent loss of expression in invasive carcinomas suggests a role in genetic progression. Am J Surg Pathol 2000, 157:755-761.
- Lynch HT, Brand RE, Lynch JF, Fusaro RM, Smyrk, TC, Goggins M, Kern SE. Genetic counseling and testing for germ-line p16 mutations in two pancreatic cancer-prone families: Case Report. Gastroenterol 2000; 119:1756-60.
- Argani P, Shaukat A, Kaushal M, Wilentz RE, Su GH, Sohn TA, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Differing rates of loss of DPC4 expression and of p53 overexpression among carcinomas of the proximal and distal bile ducts: evidence for a biologic distinction. Cancer 2001; 91:1332-1341
- Montgomery E, Goggins M, Zhou S, Argani P, Wilentz RE, Kaushal M, Booker S, Romans K, Bhargava P, Hruban RH, Kern SE. Nuclear localization of Dpc4 (Madh4, Smad4) in colorectal carcinomas and relation to mismatch repair/transforming growth factor-b receptor defects. Am J Pathol 2001; 158:537-542.
- Jones J, Hempen PM, Song J, Hruban RH, Kern SE. Detection of mutations in the mitochondrial genome in pancreatic cancer offers a "mass"-ive advantage over detection of nuclear mutations. Cancer Res 2001; 61:1299-1304.
- Ryu B, Song J, Hruban RH, Kern SE. Frequent germline deletion polymorphism of chromosomal region 8p12-p21 detected as a recurrent homozygous deletion in human tumors. Genomics 2001; 72:108-112.
- Tersmette AC, Petersen GM, Offerhaus GJA, Falatko FC, Goggins M, Rozenblum E, Wilentz RE, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Increased risk of incident pancreatic cancer among first-degree relatives of patients with familial pancreatic cancer. Clinical Cancer Res 2001; 7:738-744.
- Ryu B, Jones J, Hollingsworth MA, Hruban RH, Kern SE. Invasion-specific genes in malignancy: SAGE comparisons of primary and passaged cancers. Cancer Res 2001; 61:1833-1838.
- Su GH, Bansal R, Montgomery E, Yeo CJ, Hruban RH, Kern SE. ACVR1B (ALK4) gene mutations in pancreatic carcinoma. Proc Natl Acad Sci USA 2001; 98:3254-3257.
- McCarthy DM. Brat DJ, Wilentz RE, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Pancreatic intraepithelial neoplasia and infiltrating adenocarcinoma: analysis of progression and recurrence by DPC4 immunohistochemical labeling. Hum Pathol 2001; 32:638-642.
- Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan S, Leach SB, Ryu B, Goggins M, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Discovery of new markers of cancer through serial analysis of gene expression (SAGE): prostate stem cell antigen (PSCA) is overexpressed in pancreatic adenocarcinoma. Cancer Res 2001; 61:4320-24
- Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra D, Klöppel G, Longnecker D, Lüttges J, Offerhaus GJA. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol 2001; 25:579-586.
- Sohn TA, Su GH, Ryu B, Yeo CJ, Kern SE. High-throughput drug screening of the DPC4 tumor-suppressor pathway in human pancreatic cancer cells. Ann Surg 2001; 233:696-703.
- Feldser DM, Kern SE. Oncogenic functionality of the dinucleotide KRAS2 mutations: G12F and GG12-13VC. Human Mutation 2001; 18:357.
Reviews, Editorials, and Chapters ▼
- Kern SE. Clonality: More than just a tumor-progression model. (Editorial) J Natl Can Inst 1993; 85:1020-1021.
- Kern SE. p53: Tumor suppression through control of the cell cycle. (Editorial) Gastroenterology 1994; 106:1708-1711.
- Kern SE. Oncogenes/proto-oncogenes and tumor suppressor genes in human neoplasia. In Cellular and Molecular Pathogenesis. Sirica AE, ed. Raven Press 1996. pp 321-340
- Yeo CJ, Hruban RH, Kern SE, Abrams RA, Grochow LB, Griffin CA, Cameron JL. Adenocarcinoma of the pancreas: Factors influencing outcome following pancreaticoduodenectomy - The Johns Hopkins Experience. (Review) The Cancer Bulletin 1994; 46:504-510.
- Caldas C, Kern SE. K-Ras mutation and pancreatic adenocarcinoma. (Review) Int J Pancreatol 1995; 16:192.
- Hahn SA, Kern SE. Molecular genetics of exocrine pancreatic neoplasms. (Review) Surg Clin N Amer 1995; 55:857-869.
- Hahn SA, Kern SE, Schmiegel W-H. Molekularbiologische Veränderungen bei gastrointestinalen Tumoren, Diagnostische und therapeutische Perspektiven. Deutsches Ärzteblatt 1995; 92:137-143.
- Schutte M, Kern SE. The molecular genetics of pancreatic adenocarcinoma. (Chapter) In Pancreatic Cancer: Molecular and Clinical Advances. Neoptolemos and N. Lemoine, eds., Blackwell Scientific, Oxford, 1996. pp 115-132.
- Lynch HT, Smyrk T, Kern SE, Hruban RH, Lightdale CJ, Lemon SJ, Lynch JF, Fusaro LR, Fusaro RM, Ghadirian P. Familial pancreatic cancer: A review. Seminars in Oncology 1996; 23:251-275.
- Yeo CJ, Kern SE, Hruban RH, Camerson JL. Pancreatic cancer: New aspects of genetics and surgical management: The Johns Hopkins experience. Asian J Surg 1997; 20:221-228.
- Moskaluk CA, Kern SE. Molecular genetics of pancreatic cancer. (Chapter) In Advances in Pancreatic Cancer, H Reber, ed., The Human Press, 1998. pp 3-20.
- Moskaluk CA, Kern SE. Mad About Cancer: DPC4 and Other TGF-b Pathway Genes in Human Cancer (Review) BBA (Reviews on Cancer) Online 1996; 1288: M31-M33.
- Goggins M, Hruban RH, Kern SE. Hereditary Pancreatic Cancer - Part I: Genetic profile of the disease. Oncology News International 1997; 6 (6):24. Hereditary pancreatic Cancer - Part II: The candidate genes. Oncology New International 1997; 6 (7):8. Hereditary Pancreatic Cancer - Part III: Clinical recognition of hereditary predisposition. Oncology News International 1997; 6 (8):15.
- Hruban RH, Yeo SJ, Kern SE. Screening for pancreatic cancer. In Cancer Screening: Theory and Practice, B. Kramer, P. Provok, and J. Gohagan, eds., Dekker Publ, 1999, pp 441-459
- Kern SE. Advances from genetic clues in pancreatic cancer. Curr Opin Onc 1998; 10:74-80.
- Hruban RH, Petersen GM, Ha P, Kern SE. Genetics of pancreatic cancer: From genes to families. Surg. Onc. Clin N Amer 1998; 7:1-23.
- Slebos RJC, Ceha HM, Kern SE, Hruban RH. Molecular genetics of pancreas cancer. Dugan M and Sarkar F, eds. BioTechniques Books 1998, pp 65-82.
- Hahn SA, Kern SE, Schmiegel W-H. Neue molekularbiologische Erkenntnisse aus des Pankreaskarzinom-Forschung, Diagnostische und therapeutische Perspektiven. Deutsches Ärzteblatt 1997; 94:3-10.
- Hruban R, Offerhaus GJA, Kern SE, Goggins M, Wilentz RE, Yeo CJ. Tumor-suppressor genes in pancreatic carcinoma. (Review) J Hep Bil Pancr Surg 1998; 5:383-391.
- Hilgers W, Kern SE. The molecular genetic basis of pancreatic cancer. (review) Genes Chrom Cancer 1999; 26:1-12.
- Goggins M, Kern SE, Offerhaus GJA, Hruban RH. Progress in cancer genetics: Lessons from pancreatic cancer. Annals of Oncology 1999; 10 Suppl 4: S4-S8.
- Hruban RH, Wilentz RE, Goggins M, Offerhaus GJA, Kern SE. Pathology of incipient pancreatic cancer. Annals of Oncology 1999; 10 Suppl 4: S9-S11.
- Hruban RH, Petersen GM, Goggins M, Termette AC, Offerhaus GJA, Falatko F, Yeo CJ, Kern SE. Familial pancreatic cancer. Annals of Oncology 1999; 10 Suppl 4: S69-S73.
- Hruban RH, Goggins M, Kern SE. Molecular genetics and related developments in pancreatic cancer. (Review) Curr Opin Gastro 1999; 15:404-409.
- Klöppel G, Hruban RH, , Longnecker DS, Adler G, Kern SE, Partanen TJ. Ductal adenocarcinoma of the pancreas. World Health Organization. 2000, pp 219-230.
- Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res 2000; 6:2969
- Kern SE. Molecular genetic alterations in ductal pancreatic adenocarcinomas. (review) Med Clin N Amer 2000; 84:691-695.
- Su GH, Kern SE. The molecular genetics of pancreatic ductal neoplasia. (review) Curr Opin Gastro 2000; 16:419-425.
- Hruban RH, Wilentz R, Kern SE. Genetic progression in the pancreatic ducts. (Commentary) Am J Pathol 2000; 156:1821-1825.
- Hruban RH, Yeo CJ, Kern SE. Pancreatic cancer. (Chapter) In Metabolic and Molecular Basis of Inherited Diseases (MMBID): The Genetic Basis of Human Cancer, B Vogelstein and KW Kinzler, section eds., McGraw-Hill, CD-ROM edition, 1997 and In The Genetic Basis of Human Cancer, , B Vogelstein and KW Kinzler, eds., McGraw-Hill, 7th edition, 1998, pp 603-614 and 8th edition, 2001, pp 1077-1090.
- Hruban RH, Offerhaus GJA, Kern SE. Familial pancreatic cancer. In Atlas of Clinical Oncology: Pancreatic Cancer. John L. Cameron, ed. American Cancer Society, 2001, pp. 25-36.
- Kern SE, Hruban RH. The molecular genetics of adenocarcinoma of the pancreas. In Atlas of Clinical Oncology: Pancreatic Cancer. John L. Cameron, ed. American Cancer Society, 2001, pp. 13-24.
- Kern SE, Hruban RH, Hollingsworth MA, Brand R, Adrian TE, Jaffee E, Tempero MA. A "white paper": The product of a pancreas cancer think tank. Cancer Res 2001, 61:4923-32.
- The Pancreatic Cancer NIH Progress Review Group, Tempero M and Kern SE, co-chairs. Pancreatic Cancer: An Agenda for Action. NCI publication 2001.
- Hruban RH, Iacobuzio-Donahue C, Wilentz RE, Goggins M, Kern SE. The molecular pathology of pancreatic cancer. Cancer Journal 2001; 7:251-258.
- Sohn TA, Su GH, Ryu B, Yeo CJ, Kern SE. High-throughput drug screening of the DPC4 tumor-suppressor pathway in human pancreatic cancer cells. Ann Surg 2001; 233:696.
- Kern SE. Progressive genetic abnormalities in human cancer. In Molecular Basis of Cancer, 2nd edition, J. Mendelsohn, M. Israel, L. Liotta, P.M. Howley, eds. W.B. Saunders Co., 2nd edition, 2001, pp 41-69.
About Scott Kern, M.D.

Laboratory for Pancreatic Cancer Research
Scott E. Kern, M.D., the inaugural Everett and Marjorie Kovler Professor of Pancreas Cancer Research, is professor of oncology at the Johns Hopkins University School of Medicine. He is also the co-director of the Gastrointestinal Oncology Program at the Sidney Kimmel Comprehensive Cancer Center.
Dr. Kern has over 20,000 citations of his work among scientific publications, the majority concerning his pioneering studies of pancreatic cancer. His publications and citations place him among the top one percent of scientists in productivity and acclaim.
Dr. Kern’s laboratory is currently developing drug screening systems to identify promising therapies for the common adult-onset cancers based on our understanding of BRCA2 and other disrupted DNA-repair features in cancer cells. His discoveries are now the basis of a number of U.S. patents and novel and ongoing therapeutic trials for pancreatic cancer at Johns Hopkins and other centers.
Scott Kern graduated from the University of Michigan at Ann Arbor where he participated in an accelerated six-year program that combined undergraduate and medical studies. He did initial research training and his residency at Michigan and became board certified in anatomic and clinical pathology, and completed his fellowship in pathology and molecular genetics of gastrointestinal cancer at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins.