The multidisciplinary team at Johns Hopkins is dedicated to providing the best cutting-edge care to our patients with neuroendocrine tumors of the pancreas.
Neuroendocrine tumors run in some families (called "familial" or "hereditary"), and some produce hormones that can cause symptoms (called "functional" or "syndromic"). In addition, neuroendocrine tumors can arise in many different organs, and some neuroendocrine tumors are aggressive, while others are relatively slow growing. Simply put, the complexity of managing patients with a neuroendocrine tumor requires the multidisciplinary expertise at centers such as Johns Hopkins.

Our Team
Our team includes highly-experienced surgeons, radiologists, interventional radiologists, pathologists, medical oncologists, radiation oncologists, gastroenterologists and geneticists is supported by a dedicated team of nurses, nurse practitioners and physician assistants. Members of our team are recognized as international experts in their fields, and the group at Hopkins emphasizes teamwork in their approach to patient care.
One thing that is special about the multidisciplinary team approach at Johns Hopkins is that it is not a "one-size-fits-all" approach. Instead, we recognize that no two patients with a neuroendocrine tumor are alike, and we treat the patient who has a tumor, not just the tumor in isolation. Most importantly, we care about, and are dedicated to, our patients.
About Neuroendocrine Tumors
Neuroendocrine tumors are treated very differently from other cancers in the body. The cells of neuroendocrine tumors are recognized as being "neuroendocrine" (having neuroendocrine differentiation) because they either produce a hormone or because the cells of the tumor contain small granules (called neuroendocrine granules) that are characteristic of normal cells that produce hormones.
Neuroendocrine tumors are important to recognize for four reasons:
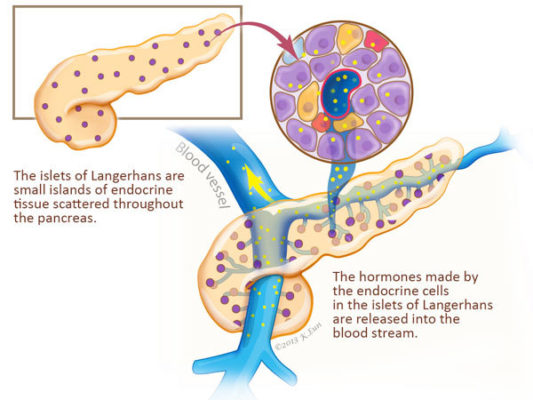
1. Some neuroendocrine tumors are functional (they produce hormones)
Many, but not all, neuroendocrine tumors produce hormones and release these hormones into the blood stream. Neuroendocrine tumors that produce hormones are called "functional" or "syndromic."
Syndromic neuroendocrine tumors are those neuroendocrine tumors that cause clinical signs and symptoms because they produce and release a hormone into the blood stream. Hormones are chemicals made by cells that can stimulate other cells in the body into action. These hormones produced by neuroendocrine tumors can cause significant symptoms and complications that have to be managed. For example, “insulinoma” is a neuroendocrine tumor of the pancreas that, as the name suggests, releases the hormone insulin into the blood stream. The insulin released by insulinomas can act on other cells in the body to greatly lower blood sugar (glucose) levels.
2. Some Neuroendocrine Tumors Run in Families
Many, but not all, neuroendocrine tumors arise as a part of an inherited genetic predisposition. Neuroendocrine tumors that are caused by an inherited genetic predisposition are called familial. In these syndromes a person inherits a defective copy a a neuroendocrine-tumor causing gene from one parent. For example, individuals with the “multiple endocrine neoplasia, type 1 (MEN 1)” syndrome inherit a genetic change in the gene MEN1 that predisposes them to develop neuroendocrine tumors of the pancreas, parathyroid glands, pituitary and upper gastrointestinal tract. These inherited genetic predispositions are important to recognize because they have implications for other family members (they too may be at risk), and because neuroendocrine tumors can arise in more than one organ, and all of the at-risk organs should be monitored.
The management of patients with a neuroendocrine tumor therefore includes a good understanding of the role that family cancer history can play in these diseases.
3. Neuroendocrine Tumors are Diagnosed Differently from Other Tumors of the Pancreas
The tools and tests used to diagnose neuroendocrine tumors are slightly different from those to diagnose the more common forms of pancreatic cancer. The specific tests can be used to diagnose neuroendocrine tumors range from specific blood tests that detect the hormones released by neuroendocrine tumors, to specific imaging tests that take advantage of the differences between neuroendocrine tumors and other tumor types.
4. Neuroendocrine Tumors are Treated Differently from Other Tumors of the Pancreas
Neuroendocrine tumors are treated in very specific ways. Because some neuroendocrine tumors grow slowly, many neuroendocrine tumors are treated surgically. Other therapies specifically target unique receptors made by neuroendocrine tumors.
News & Updates
Defining predictors of outcome in patients with a pancreatic neuroendocrine tumors.
July 29, 2025
In a real “tour de force” the team at Johns Hopkins followed over 900 patients who had a well-differentiated pancreatic neuroendocrine tumor surgically resected at Hopkins. Patients were followed for a total of 7882 person-years, and all of the clinical and pathological features of these patients were carefully reviewed by the team. This allowed the team to precisely define the features that predict outcome for patients with a pancreatic neuroendocrine tumor. The results, published in the journal Endocrine Pathology, will serve as an invaluable resource for physicians as they decide which treatments are best for their patients. For example, the authors show that virtually all patients with small (less than 2 cm) surgically resected well-differentiated pancreatic neuroendocrine tumor are cured. They also found that the rate at which the tumor cells are dividing, called the proliferation or Ki-67 rate, is an important prognostic factor.
Determining the proteins and glycoproteins made in well-differentiated pancreatic neuroendocrine tumors.
July 29, 2025
In the April 2025 issue of the journal Cancer Cell, a team of scientists in China, with assistance from Dr, Hruban at Johns Hopkins, report the near complete DNA, RNA and protein-level analysis of a series of well-characterized pancreatic neuroendocrine tumors. The researchers report three proteins that may be prognostically significant, and they report potential novel therapeutic targets. This study will form an invaluable resource for other scientists studying this tumor type.
News Archive ▼

FDA Approves Lutetium For the Patient of Neuroendocrine Tumors
January 2018
In an exciting advance, the FDA today approved Lutetium for the treatment of neuroendocrine tumors, including neuroendocrine tumors of the pancreas. Lutetium is a peptide receptor radionucleotide therapy. It targets somatostatin receptor positive neuroendocrine tumors. The approval is based on a randomized phase 3 clinical trial.

April 2014
In the April 2014 issue of the Journal of the American College of Surgeons, Dr. Choti and colleagues reported that the genetic change of "alternative lengthening of telomeres (ALT+)," can be used to predict the organ of origin in patients who have a neuroendocrine tumor in their liver. In some patients it can be hard to determine if a neuroendocrine tumor in the liver originated in the pancreas, or, for example, in the small bowel. Choti and colleagues used a molecular test for ALT and found that those tumors that originated in the pancreas and spread to the liver were more likely to have ALT than were tumors that arose in the small bowel and spread to the liver. Although a research test, it is hoped that one day this approach will help patients with a metastatic neuroendocrine tumor of unknown origin. PMID: 24655849
April 2014
Collaborators from Johns Hopkins and Memorial Sloan-Kettering Cancer Center reported in the April 2014 issue of the American Journal of Surgical Pathology their experience treating patients with aggressive ("poorly differentiated") neuroendocrine tumors of the pancreas. The authors studied 44 patients with poorly differentiated neuroendocrine tumors and found that they did significantly worse than did patients with well-differentiated (low grade) neuroendocrine tumors. Eighty-eight percent of the patients with poorly differentiated neuroendocrine tumors metastatic disease at presentation, and an additional 7% later developed metastases. Follow-up information was available for 43 patients; 33 died of disease, with an average survival of only 11. The 5-year survival rate was 16.1%. This study highlights the need to accurately grade neuroendocrine tumors, and suggests that patients with poorly differentiated neuroendocrine tumors need to be treated aggressively. PMID: 24503751
November 2013
In the November 2013 issue of the American Journal of Surgical Pathology, Dr. Ralph Hruban and colleagues from the team at Johns Hopkins reviewed the microscopic grading of 297 pancreatic neuroendocrine tumors. Grading of these tumors is performed microscopically and is determined by how fast the tumor cells are dividing. The team found that the combination of two methods for determining how fast the tumor cells are dividing (the "mitotic rate" and the "Ki67 labeling index") was better than either method alone. This study highlights the importance of good pathological review in the evaluation of neuroendocrine tumors. PMID: 24121170
October 2013
Dr. Joanna Law and colleagues at Johns Hopkins report a new way to localize small neuroendocrine tumors of the pancreas in the October issue of Surgical Endoscopy. In this paper Dr. Law and colleagues show that small “fiducials” placed at the time of endoscopic ultrasound can help guide surgeons to the precise location of small neuroendocrine tumors. This more precise localization has the potential for increasing the precision of surgery and reducing the amount of pancreatic tissue that needs to be resected. PMID: 23636530
May 2013
Dr. Trevor Ellison and colleagues reported in the Annals of Surgery the 26-year experience at Johns Hopkins on the surgical treatment of pancreatic neuroendocrine tumors. In this tour de force, Dr. Ellison and colleagues found that tumor grade (how fast the tumor cells are dividing) is the most important predictor of long-term outcome for patients with a neuroendocrine tumor of the pancreas. Importantly, they propose a simple prognostic tool that integrates grade and patient sex. This study reinforces the importance of tumor grade, and highlights the extraordinary experience the Hopkins team has in treating neuroendocrine tumors. PMID: 23673766
February 2013
The team at Johns Hopkins has successfully sequenced all known human genes in a series of medullary carcinomas of the thyroid. As reported in the Journal of clinical endocrinology, Nishant Agrawal and colleagues at Johns Hopkins, in a real tour de force, sequenced the exomes (all 21,000 known human genes) of a series of 17 clinically well-characterized medullary cancers of the thyroid. They then validated their findings in a series of 40 additional cancers. The authors found that the genes RET, HRAS and KRAS are frequently mutated (the DNA is altered abnormally) in these cancers. This study is important because it defines the genetic abnormalities driving the formation of medullary cancers of the thyroid. J Clin Endocrinol Metab. 2013 Feb;98(2):E364-9. PMID: 23264394
February 2013
In the February 2013 issue of the journal, The American Journal of Surgical Pathology, Dr. Chad McCall and colleagues from the Multidisciplinary Neuroendocrine Tumor Team at Johns Hopkins reported that the correct microscopic grading of pancreatic neuroendocrine tumors plays an important role in establishing the prognosis for patients. Dr. McCall and his colleagues found that it is important that pathologists use "Ki-67 stain" to assess tumor grade, in addition to the “old-fashioned” method of simply counting dividing cells (mitoses). PMID: 24121170
August 2012
Dr. Siva Raman and colleagues in Radiology at Johns Hopkins report in the August 2012 issue of the American Journal of Radiology on the imaging appearance of pancreatic neuroendocrine tumors, as well as a number of mimics on computerized tomography (CT). They demonstrate that pancreatic neuroendocrine tumors have a distinct appearance on imaging, and emphasize that experience is needed because a number of other entities can mimic the appearance of a neuroendocrine tumor radiologically. PMID: 22826391
March 2011
In an exciting breakthrough, Dr. Y. Jiao and colleagues at Johns Hopkins reported in the March 2011 issue of Science that they successfully sequenced the exomes (all 21,000 known human genes) in a series of pancreatic neuroendocrine tumors. This represents a major advance in our understanding of the gene changes that drive these tumors. Dr. Jiao and colleagues discovered a new cancer pathway (ATRX and DAXX mutations leading to the alternative lengthening of telomeres) and several gene changes (mutations) that may be targetable (treatable) with currently available drugs. PMID: 21252315